
An Open Letter to Dr. Michael Behe (Part 5)
Dear Dr. Behe
It is good to see that you agree that the Golgi targeting sequence is an example of a binding site. However, you don’t get to ignore it because “viral proteins are special”. As I showed in the post you are supposed to be replying to, this is nonsense. In your book you categorically state HIV has developed no new binding sites, the diagram on page 145 of “Edge of Evolution” has a big zero on it. Yet your own example of a binding site is the haemoglobin S mutation, a single amino acid mutation that just clumps up proteins. You don’t write in your book “HIV has evolved several binding sites, but they don’t count because they are viral-protein-host protein interactions” or “HIV has evolved several binding sites, but they don’t count because they are equivalent to the HbS mutation”, you just write zero
Which is wrong.
An again, you are inconsistent, you are perfectly happy to consider viral-protein binding to cellular protein interactions when you think there is no evidence of them evolving (the gp120-cell surface receptor binding, CXCR4 binding anyone). Still, let us accept that you will ignore any viral-protein-cell protein interaction.
Why did you ignore the viroporin section? An example of viral-protein –viral protein interaction that generates a new structure with important functional consequences. This is a direct challenge to the very heart of your argument.
But let’s return to the Glogi binding site sequence, YRKL. Yes, the precursor of that sequence can be seen in SIVcpz/gab, the ancestor of HIV-1 M. Very likely it did come about by a sequence of intermediate selectable steps, indeed this is the most likely way all binding sites come about, rather than your unrealistic, unevidenced model of multiple amino acid changes needing to occur simultaneously.
To see why, lets look at a general model of protein-protein binding; peptide binding to an important hormone receptor, the angiotensin II (AT) receptor (there are two version of this receptor, but for simplicity I will consider only one, the AT1-R).
Before going on, I’ll talk briefly about conceptualizing binding. Binding of a protein to another entity, whether another protein, a peptide of a small non-protein molecule, can be likened to a lock and key arrangement. Surface structures on one entity (the “key”, be it protein, peptide or small molecular), fit into complementary structures on to the protein (the “lock”). This model of course is too simple, firstly electrostatic charge and the “oiliness” [1] of the binding entities play as important a role as shape. Secondly, both the lock and the key are “floppy”, unlike our familiar locks and keys proteins, peptides and small molecules can flex and stretch into different shapes. This way a diverse number of molecules can bind to the same binding site (otherwise we wouldn’t have drugs, which mimic often bizarrely, natural hormones and enzyme substrates). With the image of a “floppy” lock and key in our minds, let’s proceed.
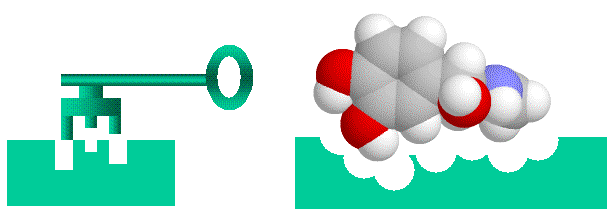 > Binding of ligands to proteins like enzymes or hormone receptors has been likened to a “lock and key” arrangement, where the molecule fits into a space in the protein in a similar manner to a key fitting into a lock. While a useful analogy to help us visualise things, this image can be misleading as we will see, as various binding interactions are important, not just physical “lumps and bumps”. Here I have used a molecule of adrenaline to illustrate the concept.
> Binding of ligands to proteins like enzymes or hormone receptors has been likened to a “lock and key” arrangement, where the molecule fits into a space in the protein in a similar manner to a key fitting into a lock. While a useful analogy to help us visualise things, this image can be misleading as we will see, as various binding interactions are important, not just physical “lumps and bumps”. Here I have used a molecule of adrenaline to illustrate the concept.
The AT1 receptor is a 359 amino acid long protein which binds the peptide angiotensin II (AII) and related peptides. AII and similar ligands bind to a specific amino-acid sequence in the AT1 receptor, DNKH in the one letter amino acid code.
D(Aspartate 281) N(Asparagine 111) K(Lysine 199) H(Histidine 256)
These amino acids are not contiguous, but separated across different loops of the protein, forming a lining to a pocket in the protein molecule.
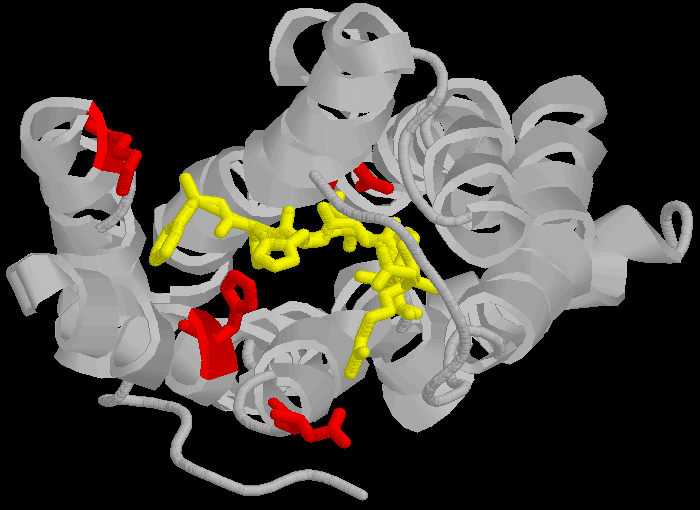 > The three dimensional cartoon structure of the AT1-receptor with AII bound. We are looking at the receptor from above, with some chains removed for clarity. The transmembrane helicies are shown as grey coils, AII is shown in yellow, the DNKH amino acids are shown in red. See http://home.mira.net/~reynella/chime/ang_tuta.htm for 3D structures (needs MDL CHIME plug-in to view and manipulate the 3D structure).
> The three dimensional cartoon structure of the AT1-receptor with AII bound. We are looking at the receptor from above, with some chains removed for clarity. The transmembrane helicies are shown as grey coils, AII is shown in yellow, the DNKH amino acids are shown in red. See http://home.mira.net/~reynella/chime/ang_tuta.htm for 3D structures (needs MDL CHIME plug-in to view and manipulate the 3D structure).
As I said above, the AT receptors bind angiotensin and similar peptides to the ligand binding site. As with the receptor, not all of the ligand sequence binds to the binding site. While here we are talking about small molecules, the principle is the same for segments of larger proteins binding to another protein. The sequences of ligands are shown in the one letter amino acid code, with the amino acids that bind being shown in bold.
| AII | DRVYIHPF |
| AIII | RVYIHPF |
|SarIle |S**R**V**Y**IHPI<br />
|SarAsp |S**D**V**Y**IHPI<br />
|CGP |NYKRHPI<br />
| AIV | VYIHPF |
Now, if you only looked at AII and the related hormone AIII, you would believe that three binding sites RYF, were required for binding. You might puzzle over how you could simultaneously get three replacements in an ancestral, non-binding peptide so that it would bind to this site.
But as we look at SarIle and SarAsp, we can see a way. These have only two of the three binding amino acids present in AII and AIII, yet they bind very well indeed. Maybe only binding to the the DN part of the DNKH motif in the AT-1R is critical. However, AIV, which dones not have a D binding amino acid, still binds (not as well as the others, but it still binds enough to be selected), so we can see selectable binding can come about more simply, and via different pathways.
It can be even simpler than that. The hormone CGP binds to completely different amino acid’s in the ligand binding cleft, not DNKH at all, so there more than one way to get to bind specifically to a something as highly selective as a hormone receptor binding site. In terms of the lock and key mechanism, your claim, Dr. Behe, is that all tines of the “key” must be in place simultaneously to fit in the lock. But as we have seen above, this is not correct, even a very simple “key” will fit into the lock (keeping in mind both the lock and key are “floppy”).
All we need to get Darwinian selection of binding sites is to have weak but selectable binding. This can be accomplished with very simple changes, and a variety of unrelated structures, even for very highly restricted structures. Note again, while this example is small peptides binding to a receptor, a small peptide is in principle the same as a surface loop of a protein binging to another protein. Your model of highly restricted sequences which have to be in position simultaneously is just wrong.
So now Dr. Behe, keep this in mind, and reflect on Wang (2002) again, and paying particular attention to the role of amino acid F43 in binding of proteins such as gp120 to CD4.
Yours sincerely
A male featherless biped named Ian Musgrave
[1] Some amino acids are charged, and will bind to other charged amino acids, some are “oily” and will bind to other oily amino acids.